
Legionnaire Disease Outbreak in New York City
Recently New York City has reported Legionnaire diseases in several areas. As of August 7, there are: 81 confirmed cases, 3 deaths and 24 currently hospitalized. Legionnaire’s disease is a type of severe pneumonia caused by Legionella bacteria, most often Legionella pneumophila.
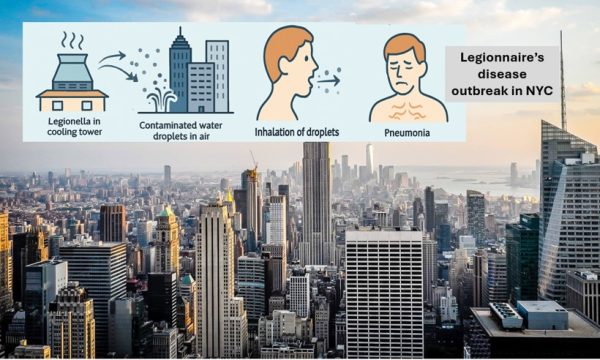
Causes: Legionella bacteria thrive in freshwater but can become a health hazard when they grow in man-made water systems like cooling towers, hot tubs, fountains, plumbing systems, and air-conditioning units for large buildings.
Spread: People get infected by inhaling small water droplets (mist) containing bacteria. It’s not spread person-to-person.
Who is at a high risk for Legionnaires’ disease?
- People who are 50 years old or older
- Smoking habit
- Suffering from chronic lung disease
- With weakened immune system
- On medicines that might weaken immune system
Why and how New York City had repeated outbreak of Legionnaire disease?
New York has some of the strictest laws in the U.S. for routine testing of cooling towers, meaning cases are more likely to be detected and reported.
- Dense urban infrastructure: Many large buildings with cooling towers, complex plumbing, and HVAC systems that can harbor Legionella.
- Aging water systems: Older pipes and water tanks can allow biofilm and bacteria to build up.
- Climate conditions: Warm, humid summers (especially July–September) create ideal bacterial growth conditions in water systems.
Symptoms:
Symptoms usually appear 2–10 days after exposure and may resemble the flu at first:
- Early signs: Fever, chills, headache, muscle aches, fatigue.
- Respiratory symptoms: Dry cough or with mucus, shortness of breath, chest pain.
- Other signs: Nausea, vomiting, diarrhea, confusion (especially in older adults).
Without treatment, it can progress quickly to severe pneumonia and even be fatal — particularly in people over 50, smokers, or those with weakened immune systems.
Treatment:
- Antibiotics: Usually macrolides (azithromycin) or fluoroquinolones (levofloxacin, moxifloxacin).
- Hospitalization: Often required for IV antibiotics, oxygen, and supportive care.
- Prognosis: Most people recover fully if treated promptly, but death rates can reach 5–10%, higher in vulnerable patients.
How is New York city taking care to stop the outbreak?
City of New York has thousands of cooling systems in proximity that need to be monitored. Also, NY had learned hard lessons from a major 2015 Bronx outbreak (138 cases, 16 deaths), which led to the strict inspection and maintenance laws now in place.
1. Detecting the cluster: Doctors and hospitals are required by law to report any case of Legionnaire’s disease to the NYC Department of Health (DOH) within 24 hours. They map cases and see if patients live, work, or recently visited the same neighborhood.
2. Environmental investigation: The DOH sends inspectors to cooling towers, building water tanks, fountains, and other aerosol-generating systems within the suspected area. Water samples are tested; maintenance records will be inspected.
3. Matching the DNA: Using molecular fingerprinting they compare bacteria from patients with bacteria found in water systems. If there’s a match, that building is officially identified as a source.
4. Immediate containment: The implicated system is shut down and disinfected using high concentrations of chlorine or other biocides.
5. Public warnings: The city issues alerts through all types of media, health websites, conferences, and advertisement from the city.
References:
- https://www.nyc.gov/site/doh/health/health-topics/legionnaires-disease.page
- https://health.mountsinai.org/blog/what-you-need-to-know-right-now-about-legionnaires-disease-in-new-york-city/
- https://www.cbsnews.com/newyork/news/legionnaires-disease-nyc-outbreak-how-to-prevent/
- https://abc7ny.com/post/harlem-legionnaires-outbreak-3-people-dead-among-67-diagnosed-july-25/17434477/
- Image credit: Free New york city manhattan cityscape Image <a href=”https://pix4free.org/”>Pix4free</a> : https://pix4free.org/ (Free for commercial use)
Author: Sumana Rao | Posted on: August 8, 2025
« Forever Chemicals PFAS Are Harmful For Pregnancy And The Baby First Case of Flesh Eating New World Screwworm Parasite Reported In USA »






